
RoseTTAFold All-Atom (RFAA): Design a Biomolecule in just three steps for Free
Reading time for this article is 03 minutes.

Introduction
Proteins are the workhorses of life, carrying out virtually every process in cells. Their incredible functional diversity stems from their intricate and beautiful 3D structures 1. Understanding and designing these molecular machines has been a major pursuit in biology and medicine. However, accurate protein structure prediction and design has long been a grand challenge 2 3 4. That's where the state-of-the-art RosettaFold All-Atom model(RFAA)5 from the Baker lab6 comes in.
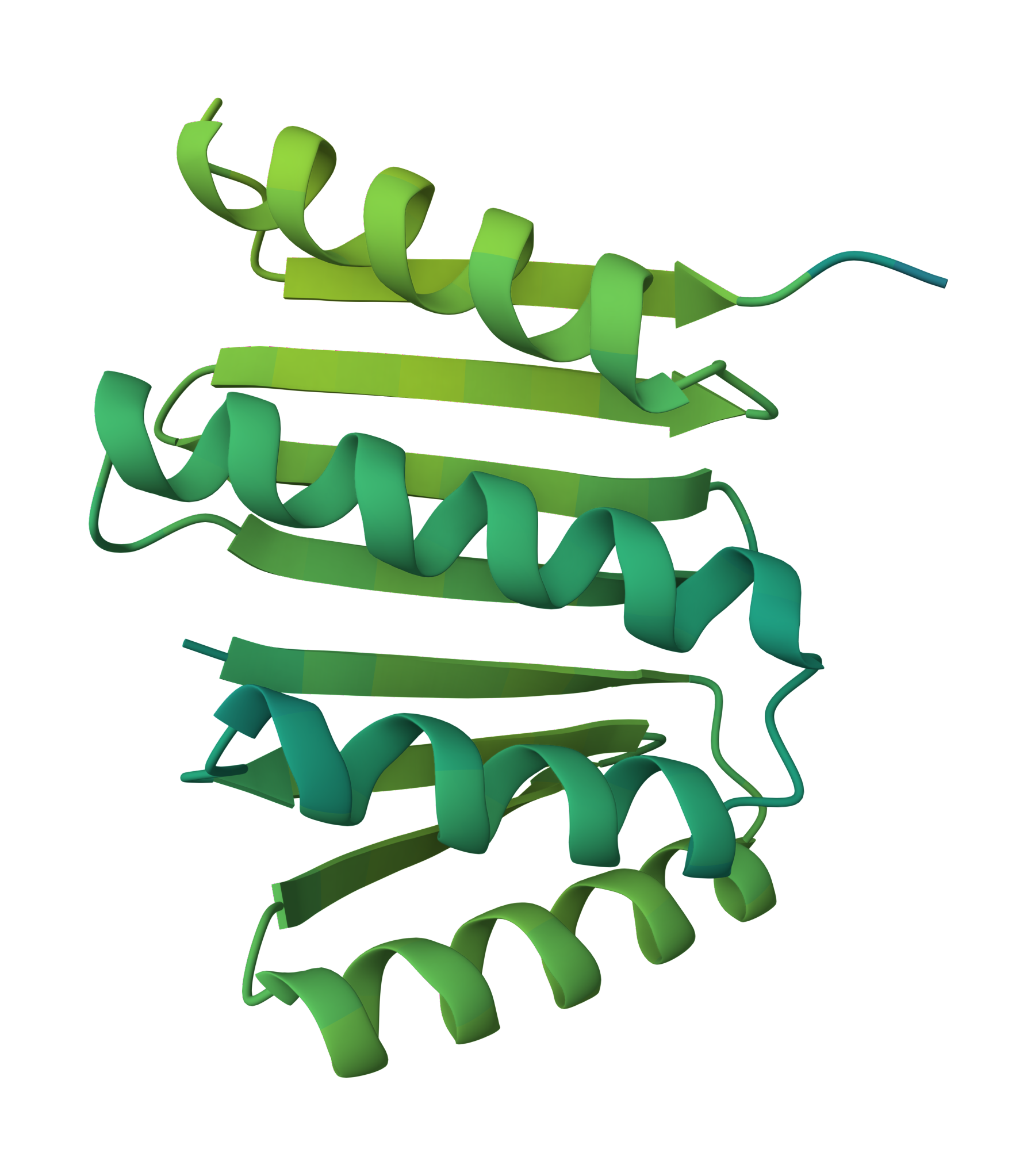
RFAA can now model a whole complex comprising any one or all such as:
Nucleic Acids (figure-2): Understanding the building blocks of genetics.
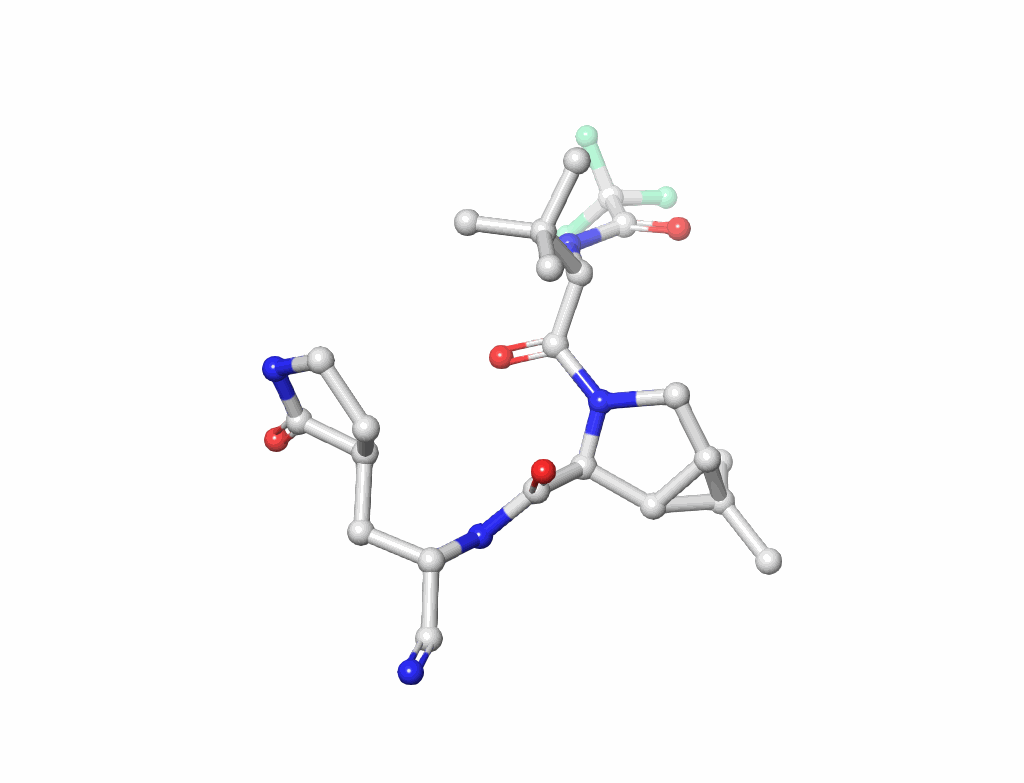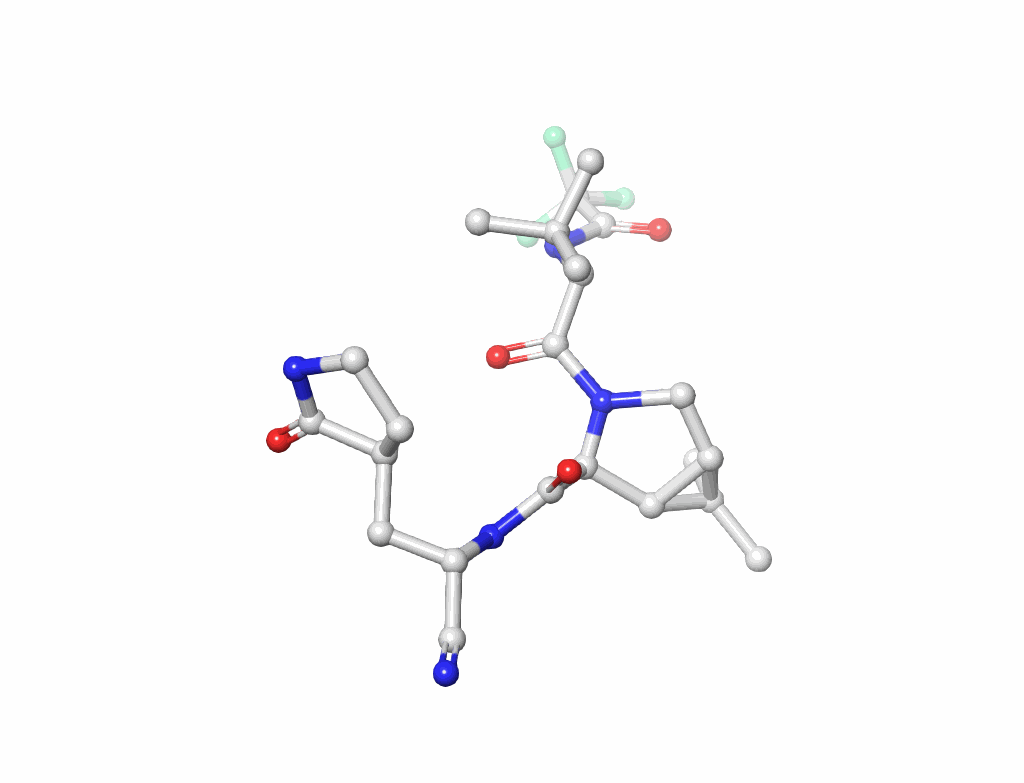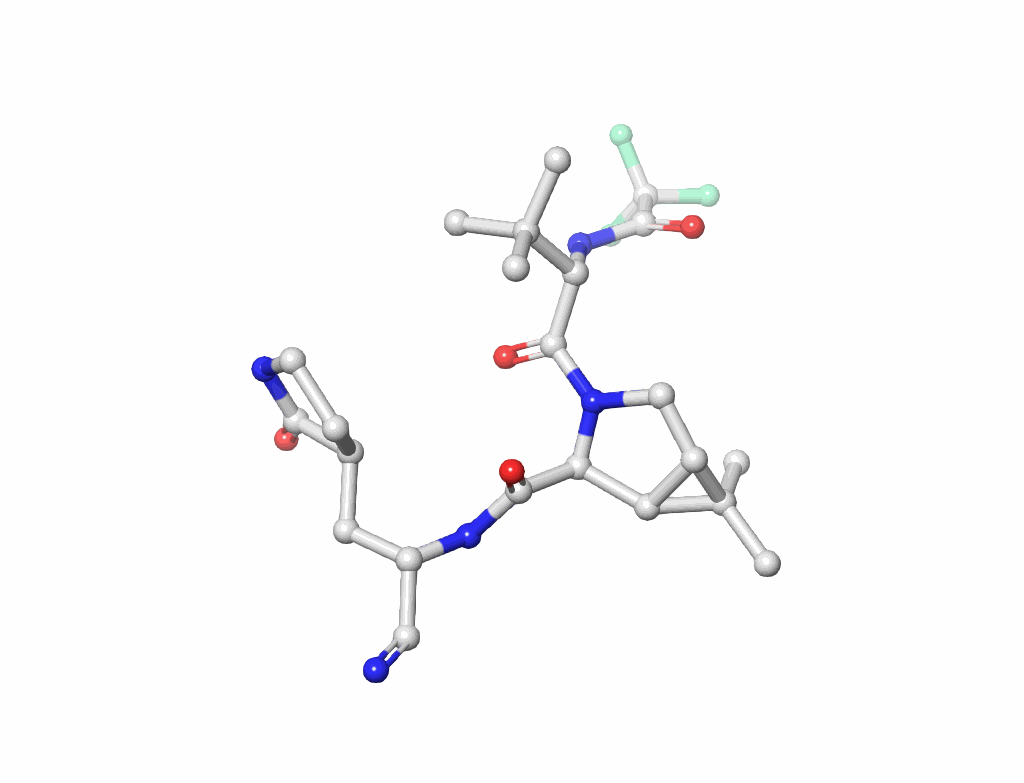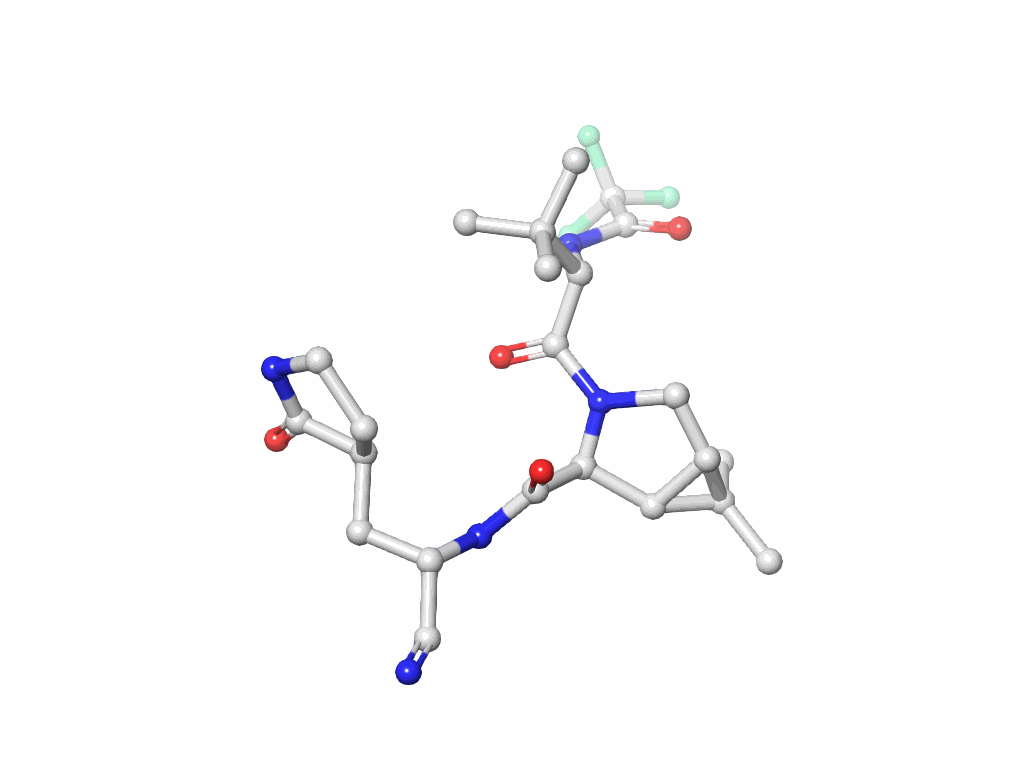
Small Molecules(figure-3): Key to drug design and interaction analysis figure 3.
Metals: Essential components in many enzymes.
Covalent Modifications: Changes that impact protein behavior.

Three things required to run RFAA!
A free sign up via google account on TamarindBio (this post is not sponsored, however we’re open for future sponsorships)
Target structure from protein data bank (PDB)
A working internet connection and a laptop or workstation.
Now Let’s design a protein-small mol drug complex
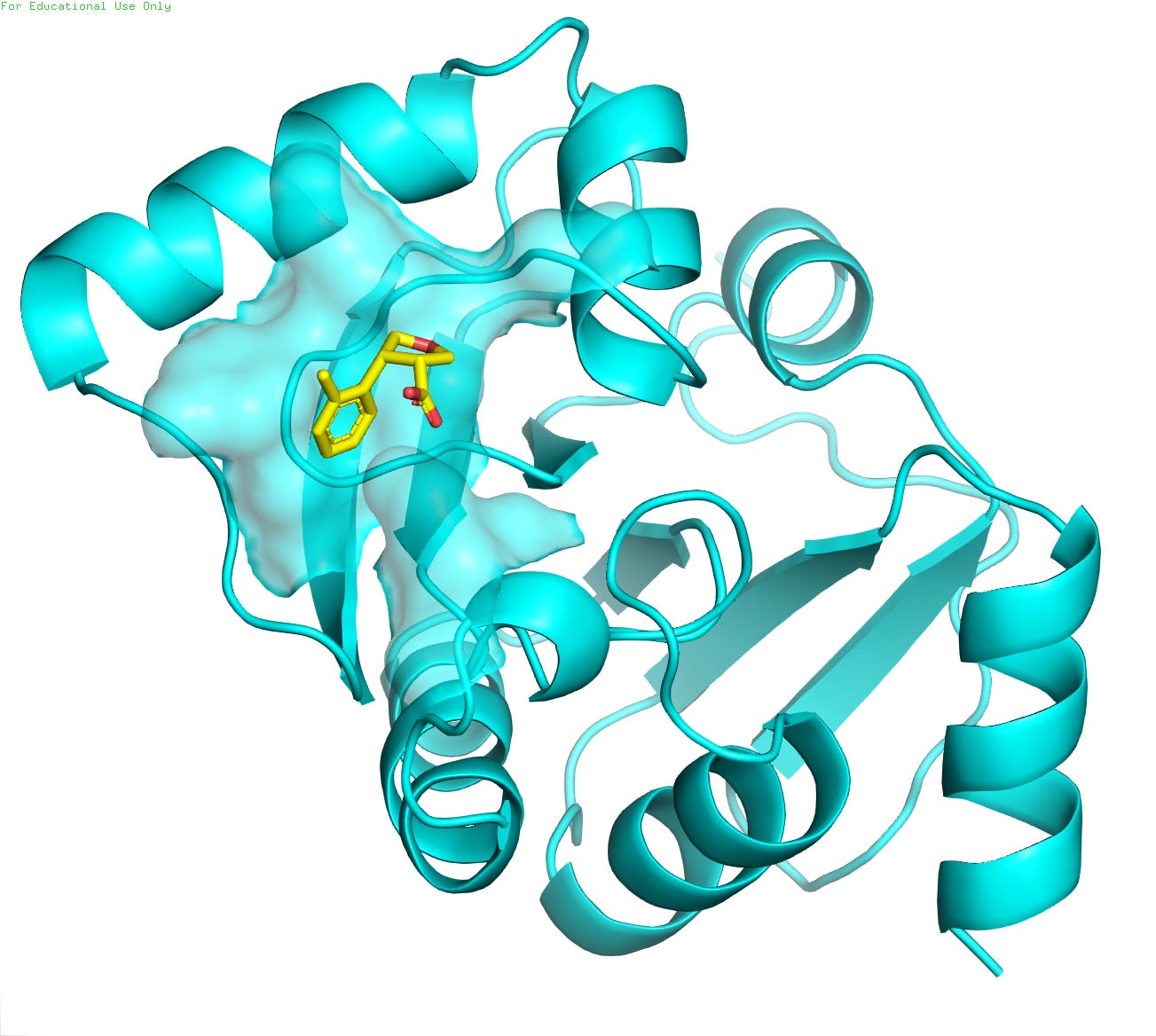
For this tutorial, we will utilize the crystal structure of the NSP3 macrodomain from SARS-CoV-2 (PDB ID: 5S3W / resolution 0.99 Å ), as depicted in Figure 4. The protein is represented in cyan, while the small-molecule ligand, a potential drug candidate, is shown in golden color. The high-resolution structural data provides invaluable insights into the molecular interactions between the viral protein and the inhibitor compound (POB0135), facilitating structure-based drug design efforts against COVID-19.
Step-1
I assume you have already sign up at Tamarind Bio and now you have logged in successfully.
Step-2
Head over to WORKFLOWS section(at top right corner).
Click the RosettaFold All-Atom
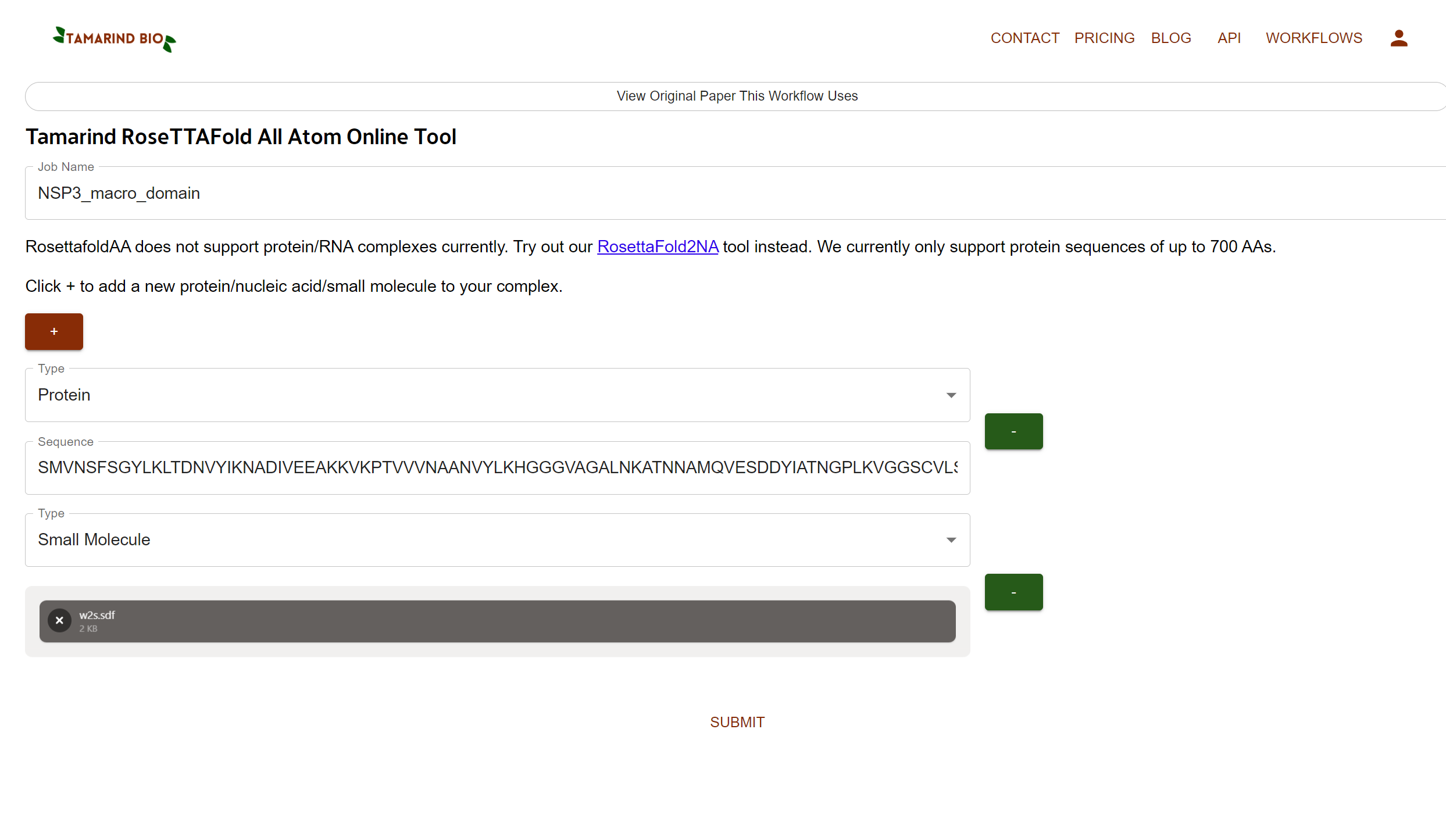
You’ll see options like a below attached screenshot
Job Name (type anything you want without space like NSP3_macro_domain)
From the Type flag we’ll select Protein, and paste the sequence in the empty area
Now we’ll Click + to add a small molecule.
Click on Drag & Drop or browse your sdf file option and upload the sdf file format.
Step-3
Submit the job and wait for the results. You’ll also get an email notification of your results.
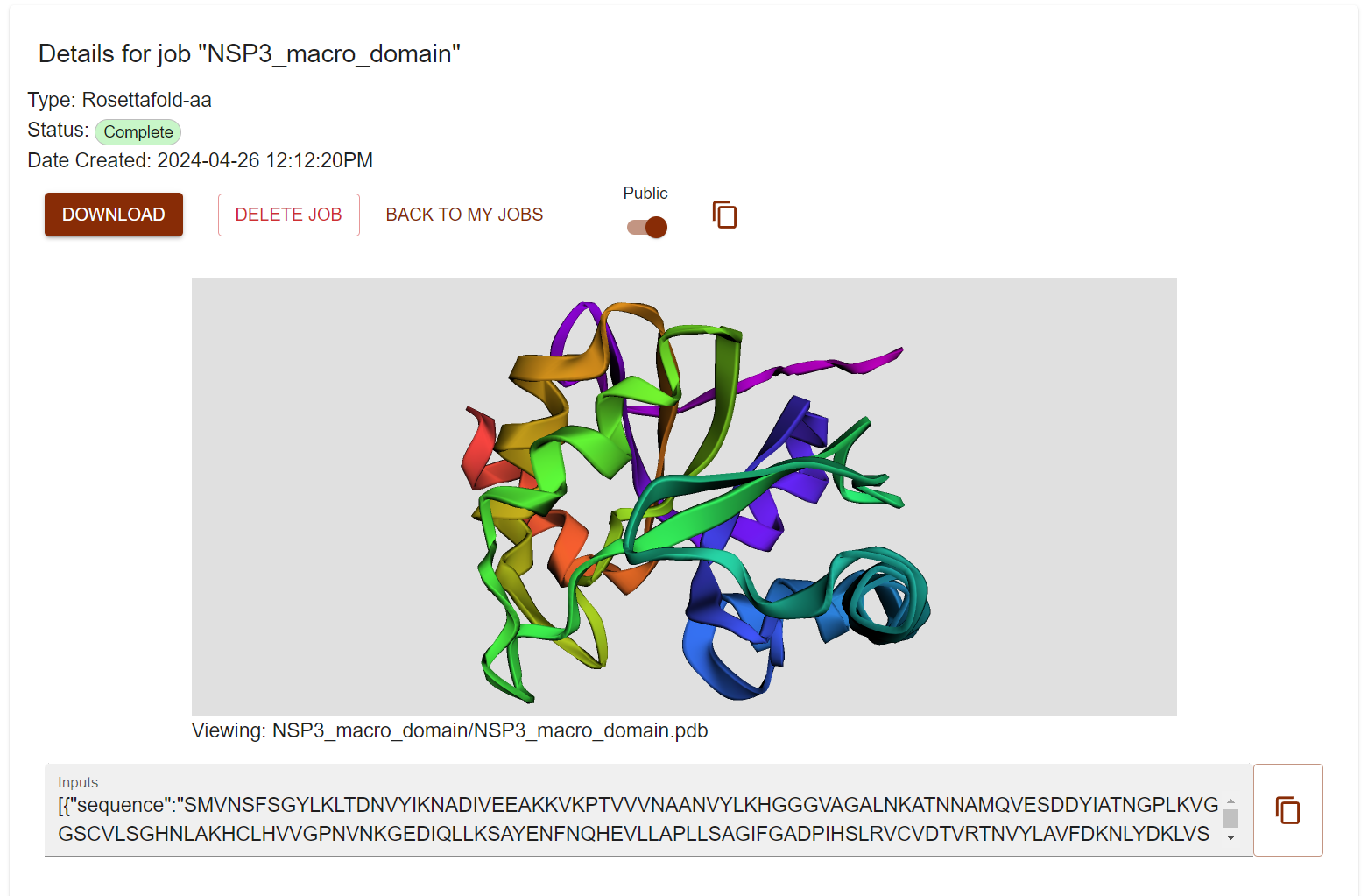
Click View and check a nice visualization of the designed structure. However I don’t see the small molecule. Don’t worry just Click DOWNLAOD and unzip the file and visualize the pdb file in pymol.
You can also public your design and anyone with link can access it freely (check it here).
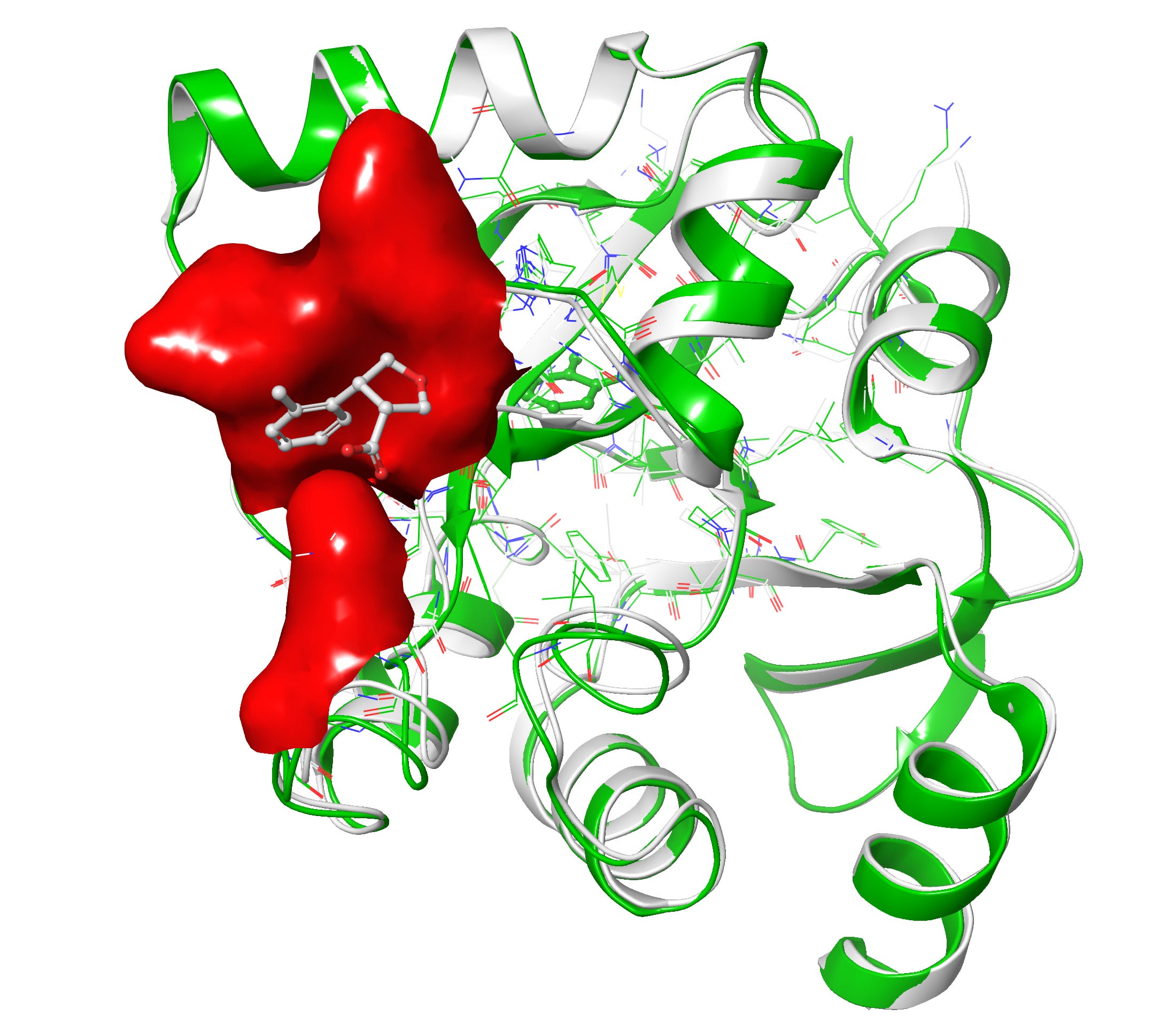
Let’s compare our designs with co-crystal
Upon superposition of crystal PDB ID: 5S3W in white and designed structure in green, I get a nice 0.7 Å rms deviation. I think RFAA did a pretty good job with the structure at figure 6-7. It also docked the small molecule in the deep pocket-center compared to co-crystal ligand on the surface in red. However upon closer look when I compared both target and design structures I get lots of clashes(bad interactions) in the designed ligand and very less in the co-crystal ligand in figure 8.
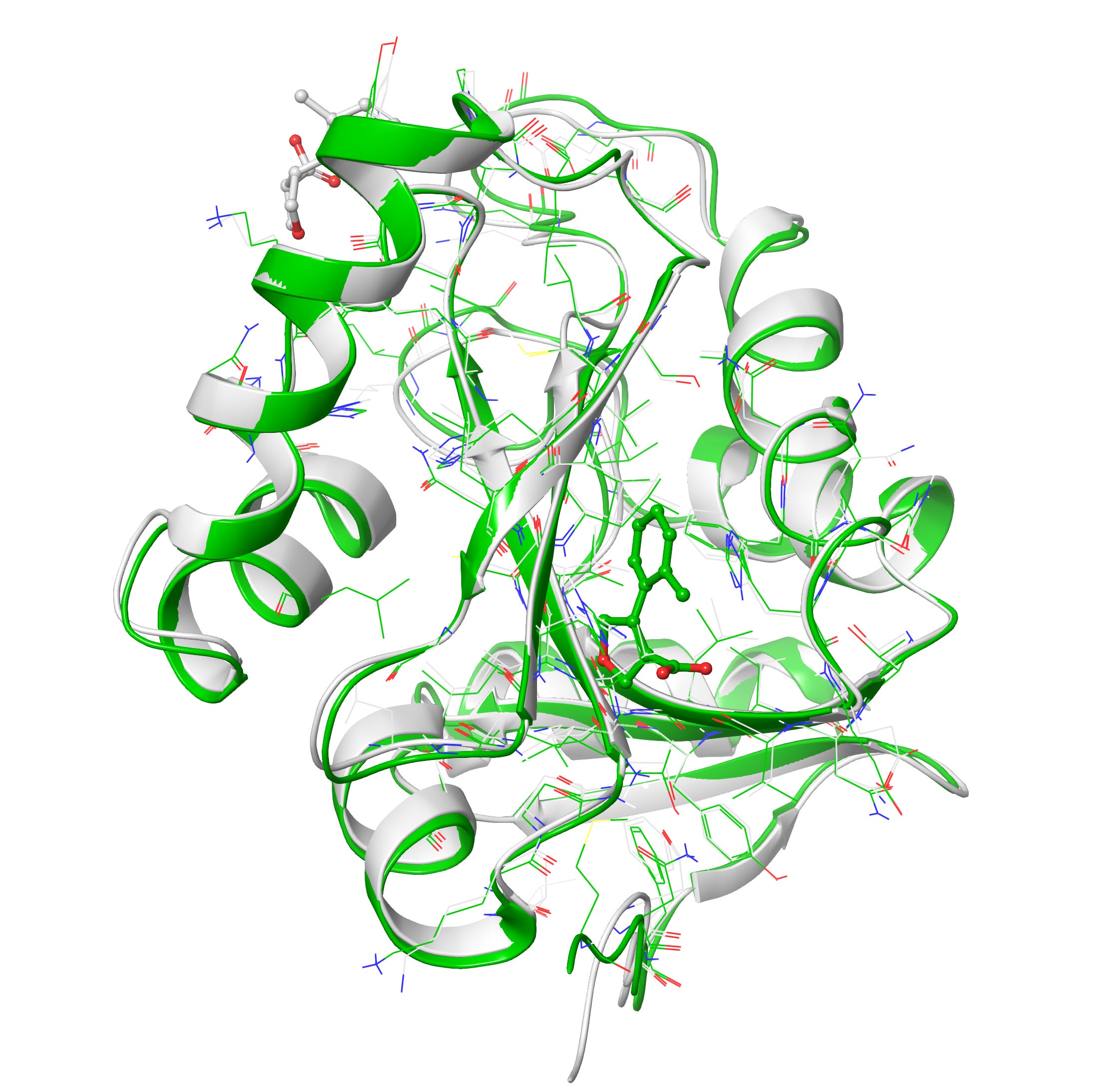
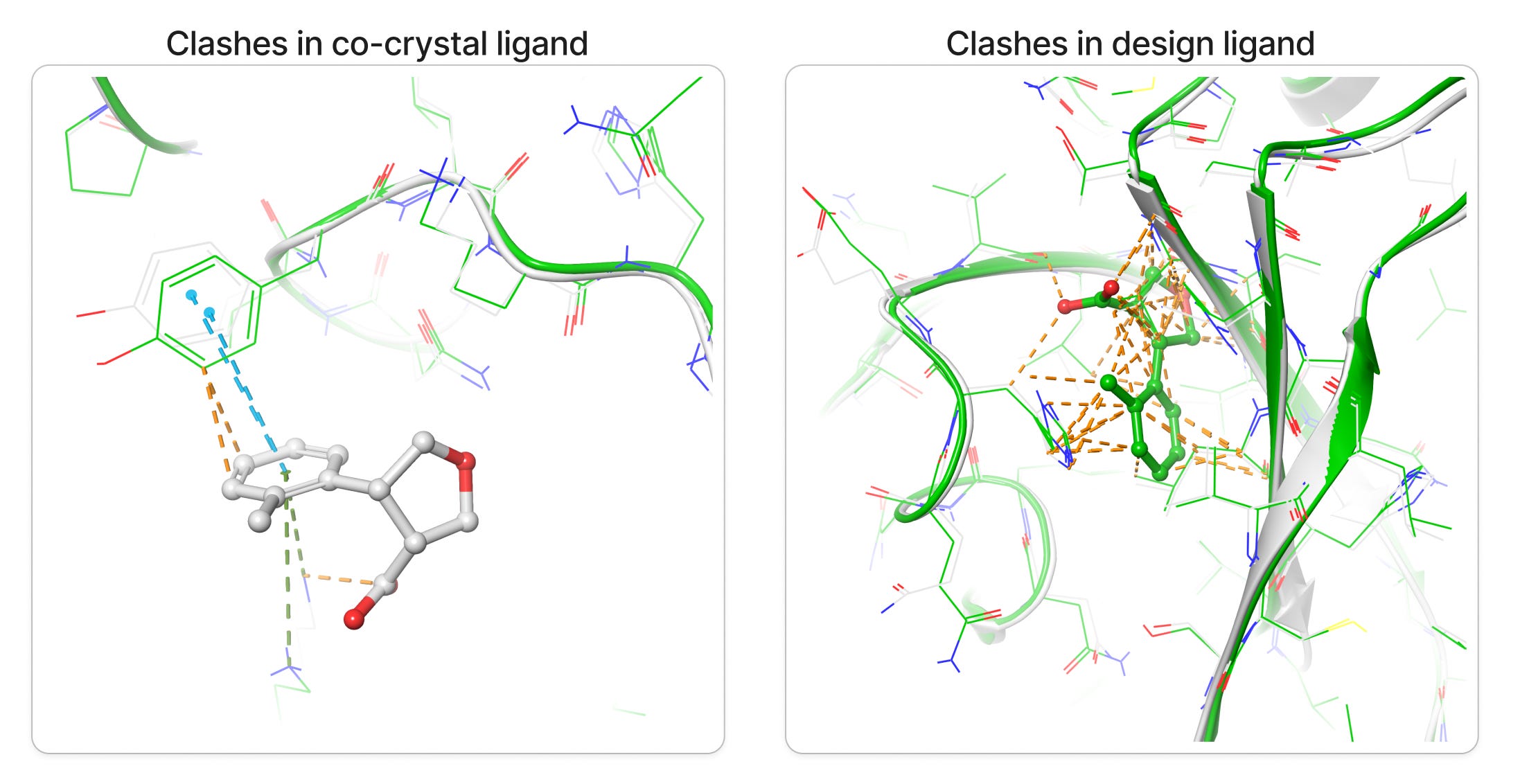
Clashes is a common problem one can get alot especially in protein and drug de novo design, luckily it can be minimized via some minimization tool also. However I prefer manually resolving the clashes by editing the structures in PyMol like If you pull it apart so that no atoms are positioned on top of each other and relax it in some energy function the minimizer will find a local minimum near to that position, which should be a configuration without clashes. Anyway I would love to hear from you on how to minimize the clashes more precisely, please do comment.
In-silico pocket prediction on the designed ligand binding site can reveal key interactions that might indicate a deeper pocket. This deeper pocket could potentially enhance the drugability of the NSP3 macrodomain. Additionally, investigating the pocket's dynamics through molecular dynamics (MD) simulations can reveal flexibility and potential allosteric effects that might influence ligand binding. Finally, experimental structure elucidation techniques like X-ray crystallography can provide high-resolution structural data to validate the in silico findings.
RFAA remain fully open source, on public code repositories like GitHub. Go fork it and play with it.
Thank you for spending your precious 05-minutes on my first blog post! I would love to hear from you about any other topic or tool discussion to include in my future posts. For any sponsorship please do not hesitate to connect via LinkedIn
https://www.nature.com/scitable/topicpage/protein-structure-14122136/
Listov, Dina, et al. "Opportunities and challenges in design and optimization of protein function." Nature Reviews Molecular Cell Biology (2024): 1-15.
Khakzad, Hamed, et al. "A new age in protein design empowered by deep learning." Cell Systems 14.11 (2023): 925-939.
Chu, Alexander E., Tianyu Lu, and Po-Ssu Huang. "Sparks of function by de novo protein design." Nature Biotechnology 42.2 (2024): 203-215.
Krishna, Rohith, et al. "Generalized biomolecular modeling and design with RoseTTAFold All-Atom." Science (2024): eadl2528.
https://www.bakerlab.org/